Whitepaper - Guideline for Biocompatibility Testing of Medical Devices
Introduction
1.1 Medical Device: The definition of "medical device" is broad; at one extreme, it can refer to a single material that can take on several physical forms, while at the other end of the spectrum, it can refer to a medical device that has numerous parts made of various materials.

What is Biocompatibility?
A wide variety of materials and components, each with distinct physical and chemical properties, make up modern medical devices. When used in items meant for general use, many of these materials pose only a minor risk, but their usage in medical devices vastly increases the range of possible safety problems. These may include the migration of chemicals from the device or the leaching of device materials as a result of heat or wear during routine use.
Adverse biological responses to implantable medical devices in patients might include everything from itchiness, soreness, or discomfort to consequences on development or reproduction to outright rejection of the device itself. Even in situations when devices have limited, transient contact with patients, like when they come in contact with the skin or internal organs and tissues during surgery, biological reactions can result in allergic reactions that can endanger the health of the patient. Additional material problems may arise from processes used in the manufacture, post-production, and use of medical devices that have a negative influence on their components and materials.
For instance, if the device comes in contact with lubricants or other chemicals while being created or maintained, its chemical integrity may be jeopardized. Sterilization and disinfection processes may lead to an increase in potentially hazardous chemical emissions, similar to prolonged use.
About ISO 10993-1:2018 and the risk management processes 10993-1:
Evaluation and Testing within a risk management Process:
The ISO 10993 series of standards currently consists of more than 20 individual standards that address various aspects of biocompatibility in medical devices. The standard ISO 10993-1:2018, Biological evaluation of medical devices – Part 1: Evaluation and testing within a risk management system are intended to address the framework of the evaluation and testing process used to assess medical device biocompatibility.
2. Evaluation of Biocompatibility of the Medical Device
The evaluation of the device, including its material components, manufacturing methods, and clinical application, including its intended anatomical location, frequency, and length of exposure, should typically be the first step in such a procedure. The potential risks from a biocompatibility perspective should be determined in light of this knowledge. Following the identification of the risks, the sponsor or expert should evaluate the information presently available on those risks and pinpoint any knowledge gaps that still exist. A strategy should be created to fill in the information gaps, either by biocompatibility testing or other assessments that effectively handle the hazards, taking into account the potential biological impact. In the proper benefit-risk context, the interpretation of the overall biocompatibility evaluation should be considered.
2.1 The evaluation must consider the advantages, disadvantages, and usability:
- Medical device configuration (such as size, geometry, and surface features), a list of the materials used in its production (qualitative), and, if applicable, the percentage and mass (in mass units) of each material.
- The composition and physical and chemical properties of the major building components.
- Any information on human exposure or clinical usage history.
- Every available toxicological and other biological safety information on the ingredients, breakdown products, and metabolites used in products and their components
- A study of pertinent recent preclinical and clinical data as well as actual testing can be included in the evaluation process. If the material has a demonstrated safe history of usage in each role and physical form that is equivalent to that of the medical device under design, such an examination can conclude that no testing is necessary.
- Annexure A of ISO 10993-1, “Endpoints to be addressed in a biological risk assessment”, provides a more detailed framework for the development of a biocompatibility assessment for medical devices, based on the category of the device and the nature of contact the device has with the body.
- Annexure B of ISO 10993-1 contains the kinds of details that can be used to show biological evaluation as a risk management practice.
- Annexure C of ISO 10993-1 details that if there is enough historical data available to do a risk evaluation of the substance and/or the medical device, testing is typically not necessary.
- Instead, the risk management process in ISO 10993-1 gives medical device manufacturers the full responsibility to:
- Thoroughly and carefully identify all potential biological risk factors unique to their device
- Evaluate the extent of the potential risk attributable to these factors
- Determine steps necessary to mitigate risks that are inconsistent with the patient health and wellbeing
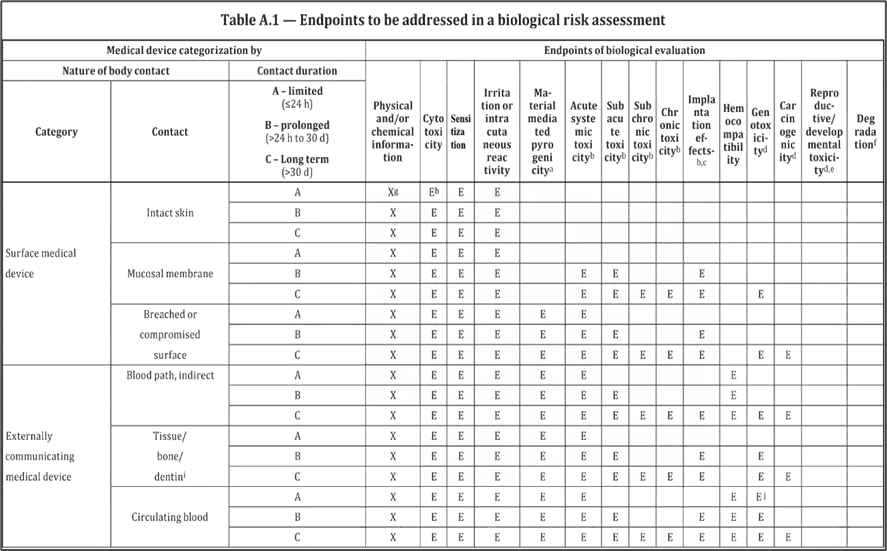
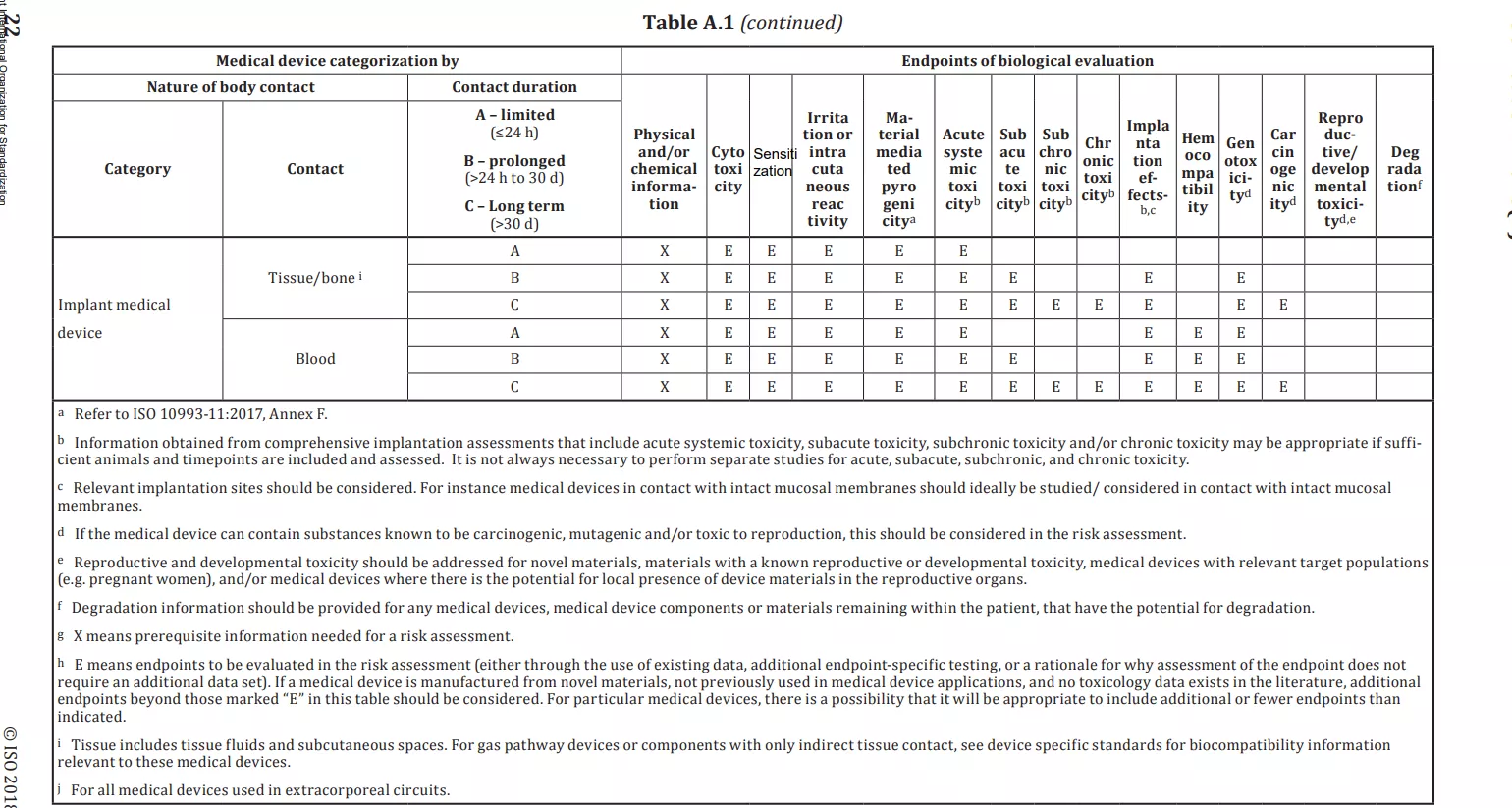
2.3 Categorization by the duration of contact
Contact duration categories medical devices shall be categorized according to the anticipated duration of contact as follows.
(A) Limited exposure: Medical devices whose cumulative sum of single, multiple, or repeated duration of contact is up to 24 h.
(B) Prolonged exposure: Medical devices whose cumulative sum of single, multiple, or repeated contact time is likely to exceed 24 h but not exceed 30 d.
(C) Long-term exposure: Medical devices whose cumulative sum of single, multiple, or repeated contact time exceeds 30 d.
Transitory-contacting medical devices
Some medical devices with limited exposure (A) have very brief/transitory contact with the body (e.g. lancets, hypodermic needles, and capillary tubes that are used for less than one minute). These devices generally would not require testing to address biocompatibility. However, for products made with materials such as coatings or lubricants that could be left in contact with body tissues after the medical device is removed, a more detailed biocompatibility assessment may be necessary.
Medical devices with multiple contact duration categories
If a material or medical device can be placed in more than one duration category, the more rigorous testing and/or evaluation considerations shall apply. With expected or intended multiple exposures to a device, the decision into which category a medical device is placed shall consider the potential cumulative effect, keeping in mind the time over which these exposures occur. If a medical device is intended to change during its lifetime, such as those that are polymerized and/or degraded in situ, the evaluation shall consider what all the different device states.
To share an example, different device states for an absorbable glue that is designed to polymerize in situ would include initial components, intermediate reaction products, completely polymerized material, and degradation products.
3. BIOLOGICAL SAFETY EVALUATION PROCESS
Medical device biological safety analysis is done in two steps:
- Biocompatibility Assessment
- Biocompatibility Report
3.1 Biocompatibility Assessment
- To comply with the criteria of the standard, device makers (or their designated contract professionals) must create a documented, written biocompatibility assessment.
- Arrangements for acquiring relevant data from published sources, internal and external data sources, and additional sources as necessary to carry out a thorough risk analysis.
- Plans for carrying out the biocompatibility assessment, including any required unique technical expertise in the medical device.
- Plans for the biocompatibility assessment and approval as part of the overall design control process.
- Arrangements for reviewing the evaluation's findings and approving any potential subsequent testing.
- Plans for the final review, approval, and documentation of the biological risk assessment's findings, including any implemented risk management measures and any residual risk.
- The characterization of the physical and chemical material properties of the device, its components, and its materials that are important to the biological safety of the device is a crucial component of the biocompatibility assessment.
A few of the elements covered by such a categorization include:
- Wear, load and fatigue, especially in load-bearing medical devices.
- Friction and the potential for associated irritation.
- Chemical interactions in material combinations in the device.
- Thermal degradation.
- Manufacturing processes that can result in environmental stress cracking, morphological changes, or degradation.
- Interactions with the anticipated use environment, such as bodily fluids and acids, and decontamination processes.
- Transportation and aging
- The chemical characterization of the device usually includes a detailed analysis of the toxicological risks posed by the device's components, as well as an examination of the types of toxic effects and the dose-response relationships associated with those effects.
The following specific toxicity endpoints should be considered while describing the chemical:
Tests need to be considered
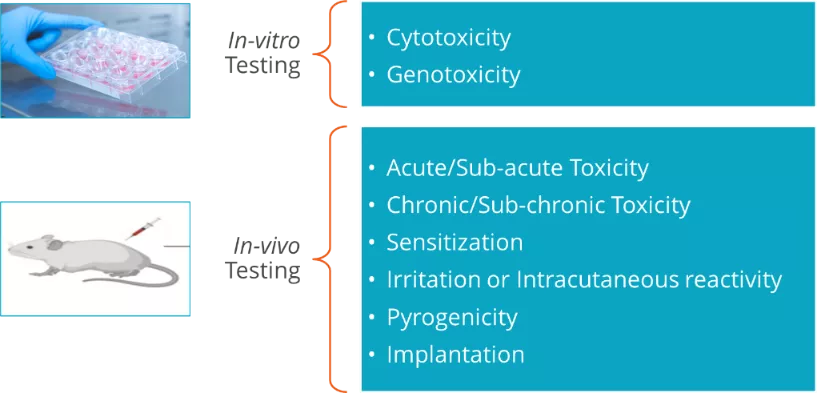
Annex A of ISO 10993-1 provides additional details on the relevant toxicity endpoints that should be considered as part of the chemical characterization process. Depending on the device category, additional testing in the following areas may also be required:
- Implantation effects
- Hemocompatibility
- Pyrogenicity
The basic 3 tests which are also referred as 3 big biocompatibility tests are
- Cytotoxicity assay
- Sensitization study
- Irritation study
A. Tests for In-vitro Cytotoxicity: ISO-10993 part-5
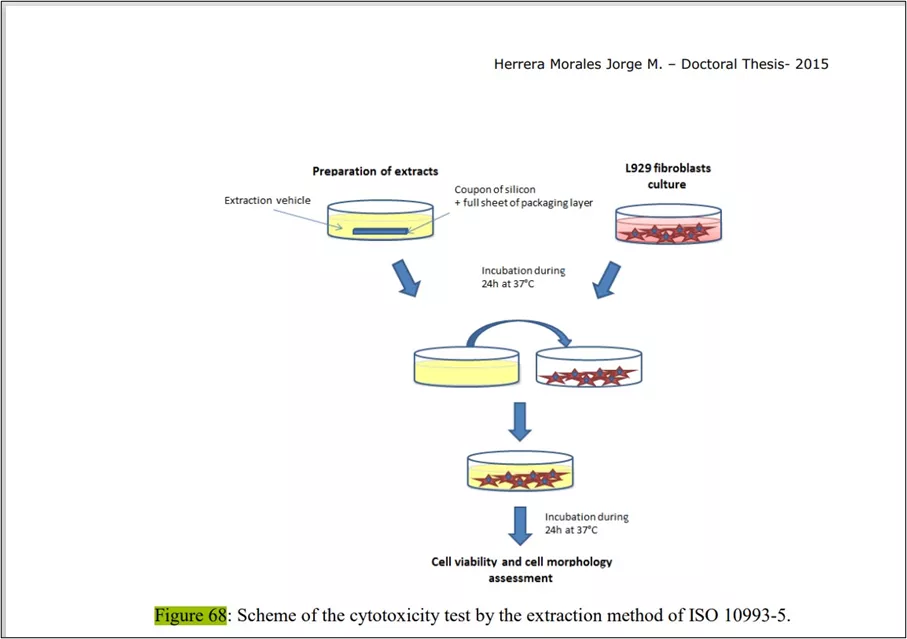
Steps involved during the experimental proc -
- To ensure the safety of the patient preliminary test needs to be performed is cytotoxicity.
- the method of performing cytotoxicity is the MTT assay
- Seed 96-well plates: 1 × 104 cells/100 µl MEM culture medium/well Incubate (37°C/5 % CO2/22 h to 26 h)
- Treat with different concentrations of test sample extract in treatment medium (100 µl) and then Incubate (37°C/5 %CO2/24 h)
- Microscopic evaluation of morphological alterations and grading according to the table
- Remove culture medium Add 50 µl MTT solution Incubate (37 °C/5 % CO2/2 h)
- Remove the MTT solution Add 100 µl isopropanol/DMSO to each well
- Detect absorption at 570 nm (reference 650 nm) in Spectrophotometer.
Steps involved during the experimental process -
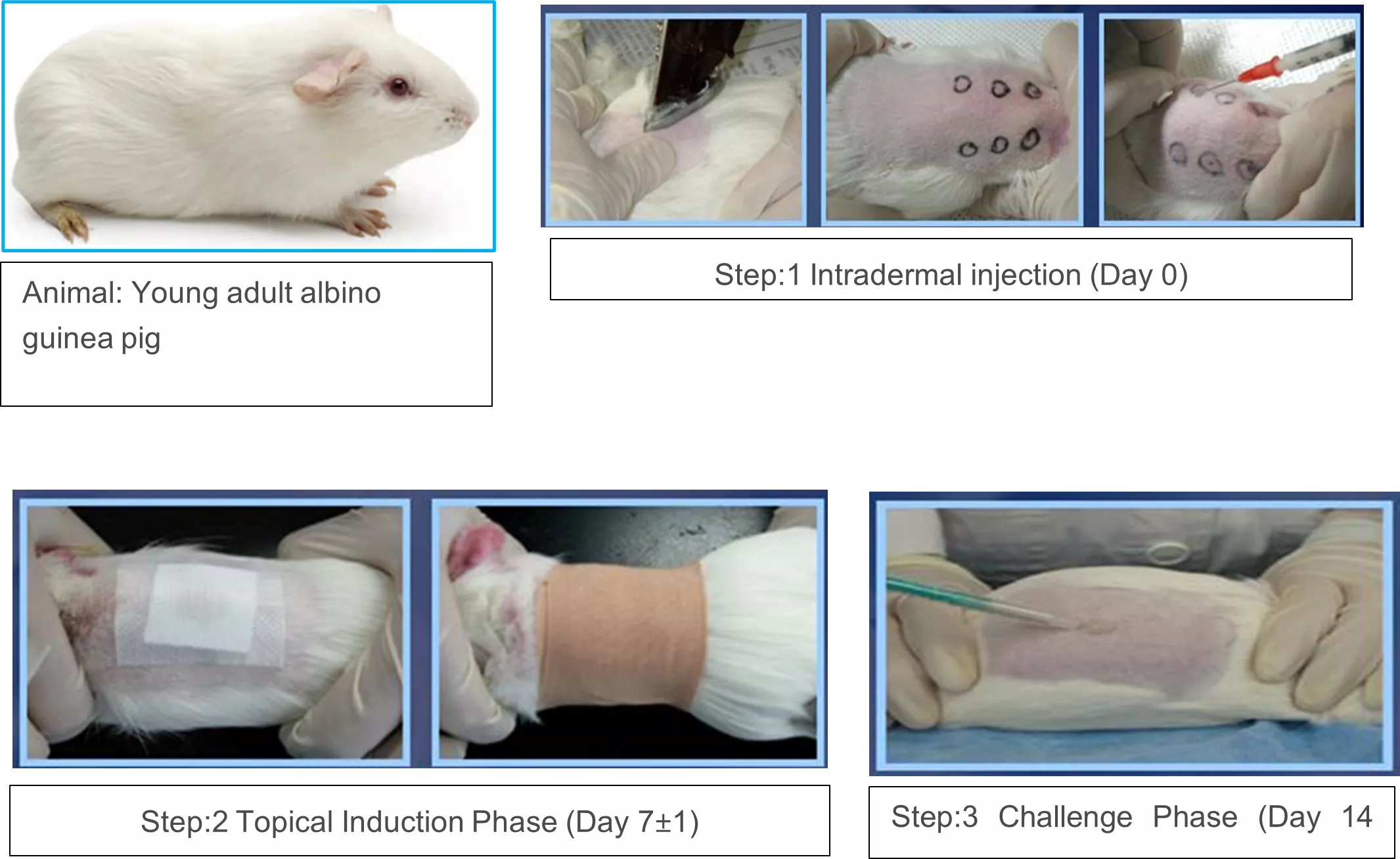
Intradermal induction phase
Make a pair of 0.1 ml intradermal injections as per ISO guidelines, for each animal, at the injection sites (A, B, and C), as shown in the figure in the clipped intra-scapular region.
Topical induction phase
At (7 ± 1) day after the completion of the intradermal induction phase, administer the test sample by topical application to the intrascapular region of each animal, using a patch of area approximately 8 cm2 (filter paper or absorbent gauze), to cover the intradermal injection sites (Figure). Remove the dressings and patches after (48 ± 2) hours.
Challenge Phase
At (14 ± 1) day after the completion of the topical induction phase, challenge all test and control animals with the test sample. Administer the test sample and a blank by topical application to sites that were not treated during the induction stage, such as the upper flank of each animal, using appropriate patches or chambers soaked in the test sample at the concentration Secure with an occlusive dressing. Remove the dressings and patches after (24 ± 2) hour.
Steps involved during the experimental process -
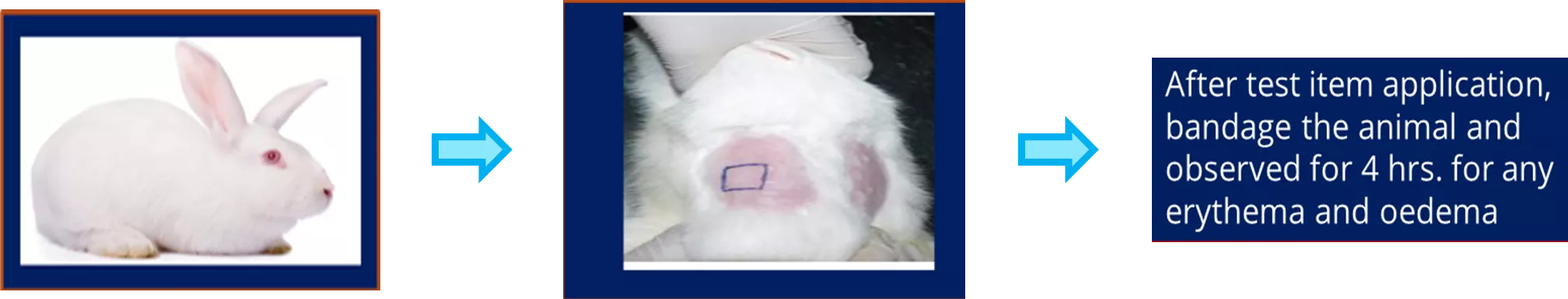
- Solid materials that have appropriate physical states (e.g. sheets, films) shall be tested without modification.
- Cover the application sites with 2,5 cm × 2,5 cm non-occlusive dressings (such as a gauze patch) and then wrap the application sites with a bandage (semi-occlusive or occlusive) for a minimum of 4 h.
- At the end of the contact time, remove the dressings and mark the positions of the sites with permanent ink. Remove residual test material by appropriate means, such as washing with lukewarm water or other suitable non-irritating solvent and careful drying.
- For single-exposure tests, record the appearance of each application site at (1 ± 0,1) h, (24 ± 2) h, (48 ± 2) h and (72 ± 2) h following removal of the patches.
- For single exposure tests, determine the primary irritation index
- (PII: Primary Irritation Index). score
0-0.4 Negligible
0.5-1.9 Slight
2-4.9 Moderate
5-8 Severe
3.2 Biocompatibility Report
The creation of the biocompatibility report is the last step in the biological safety evaluation procedure for a medical device biocompatibility report. The biocompatibility report is a crucial file when requesting that a regulatory body approve or certify a device since it supports the assertion made by the manufacturer that they have complied with ISO 10993-1 and ISO 14971's risk management standards.
- At a minimum, the Biocompatibility Report should include:
- A review of available toxicity and prior use data, pertinent to the medical device and its components.
- (Sentence repeat)Detailed information on the physical and chemical characteristics of the device and its components that come into direct or indirect contact with patients.
- Information on the processing and manufacturing process that could potentially introduce contaminants.
- Reports of all biological safety testing carried out as part of the risk assessment of the device.
- Post-production considerations and other issues.
"The processes of risk assessment are based on human judgement using the available information, supported by biological testing when appropriate," states Annex B of ISO 10993-1. As a result, it emphasises how crucial it is to review and update the biological safety evaluation of medical devices before they are put on the market to account for any new information that may emerge, such as from reports about the device's performance or safety after it has been used in a clinical setting or from subsequent biocompatibility safety literature or research.
4. SUMMARY AND CONCLUSION
In the end, very few medical procedures are risk-free. When it comes to the creation of medical devices, the dangers of their use must be weighed against any potential health advantages that a patient might receive from using them. Because of this, it's crucial to keep in mind that the results of a biological safety study must be interpreted within the proper benefit-risk framework.
The ISO 10993-1 standard offers a logical and robust framework for evaluating the biological safety of a medical device and reducing the possible danger to patients. It is based on well-established and widely accepted risk management principles. Nevertheless, it takes a team to evaluate biological safety and market medical gadgets that are both safe and efficient. Instead, a team effort between device inventors, testing facilities, toxicologists, biologists, and chemists is required for it to be successful.
Discover more
